IoT security (internet of things security)
What is IoT security (internet of things security)?
IoT security (internet of things security) is the technology segment focused on safeguarding connected devices and networks in IoT. IoT involves adding internet connectivity to a system of interrelated computing devices, mechanical and digital machines, objects, animals and people. Each thing has a unique identifier and the ability to automatically transfer data over a network. However, enabling devices to connect to the internet opens them up to serious vulnerabilities if they aren't properly protected.
The term IoT is extremely broad, and as this technology continues to evolve, the term only becomes broader. From watches to thermostats to video game consoles, nearly every technological device can interact with the internet, or other devices, in some capacity.
IoT security is even broader than IoT, resulting in a variety of methodologies falling under that umbrella. Application programming interface (API) security, public key infrastructure (PKI) authentication and network security are just a few of the methods IT can use to combat the growing threat of cybercrime and cyberterrorism rooted in vulnerable IoT devices.
Why is IoT security important?
Due to the unconventional manufacturing of IoT devices and the vast amount of data they handle, there's a constant threat of cyber attacks. Several high-profile incidents where a common IoT device was used to infiltrate and attack the larger network have drawn attention to the need for IoT security.
This article is part of
Ultimate IoT implementation guide for businesses
The ever-looming possibility of vulnerabilities, data breaches and other risks associated with IoT device usage underscores the urgent need for strong IoT security. IoT security is vital for enterprises, as it includes a wide range of techniques, strategies, protocols and actions that aim to mitigate the increasing IoT vulnerabilities of modern businesses.
IoT security issues and challenges
The more ways there are for devices to connect to each other, the more opportunities there are for threat actors to intercept them. Hypertext Transfer Protocol and APIs are just two of the channels that IoT devices rely on that hackers can intercept.
The IoT umbrella doesn't strictly include internet-based devices either. Appliances that use Bluetooth technology also count as IoT devices and, therefore, require IoT security.
The following IoT security challenges continue to threaten the financial safety of both individuals and organizations:
- Remote exposure. Unlike other technologies, IoT devices have a particularly large attack surface due to their internet-supported connectivity. While this accessibility is extremely valuable, it also gives hackers the opportunity to interact with devices remotely. This is why hacking campaigns, such as phishing, are particularly effective. IoT security, including cloud security, has to account for a large number of entry points to protect assets.
- Lack of industry foresight. As organizations continue with digital transformations, so too have certain industries and their products. The automotive and healthcare industries have expanded their selection of IoT devices to become more productive and cost-efficient. This digital revolution, however, has also resulted in a greater technological dependence than ever before. While normally not an issue, a reliance on technology can amplify the consequences of a successful data breach. What makes this concerning is that these industries are now relying on pieces of technology that are inherently more vulnerable: IoT devices. Not only that, but many healthcare and automotive companies weren't prepared to invest the amount of money and resources required to secure these devices. This lack of industry foresight has unnecessarily exposed many organizations and manufacturers to increased cybersecurity threats.
- Resource constraints. Not all IoT devices have the computing power to integrate sophisticated firewalls or antivirus software. In fact, some devices can barely connect to other devices. IoT devices that have adopted Bluetooth technology, for example, have suffered from a recent wave of data breaches. The automotive industry, once again, has been one of the markets hit the hardest.
- Weak default passwords. IoT devices often come with weak passwords, and most consumers might not be aware that they need to be replaced with more secure ones. If default passwords aren't changed on IoT devices, it can leave them vulnerable to brute-force and other hacking attacks.
- Multiple connected devices. Most households today have multiple interconnected devices. The drawback of this convenience is that, if one device fails due to a security misconfiguration, the rest of the connected devices in the same household go down as well.
- Lack of encryption. Most network traffic originating from IoT devices is unencrypted, which increases the possibility of security threats and data breaches. These threats can be avoided by ensuring all the devices are secured and encrypted.

In 2020, a cybersecurity expert hacked a Tesla Model X in less than 90 seconds by taking advantage of a massive Bluetooth vulnerability. Other cars that rely on wireless key fobs to open and start have experienced similar attacks. Threat actors have found a way to scan and replicate the interface of these fobs to steal vehicles without so much as triggering an alarm. If technologically advanced machinery, such as a Tesla vehicle, is vulnerable to an IoT data breach, then so is any other smart device.
How to protect IoT systems and devices
Enterprises can use the following tools and technologies to improve their data protection protocols and security posture:
- Introduce IoT security during the design phase. Of the IoT security risks and issues discussed, most can be overcome with better preparation, particularly during the research and development process at the start of any consumer-, enterprise- or industrial-based IoT (IIoT) device development. Enabling security by default is critical, along with providing the most recent operating systems and using secure hardware.
IoT developers should be mindful of cybersecurity vulnerabilities throughout each stage of development -- not just the design phase. The car key hack, for instance, can be mitigated by the driver placing their fob in a metal box or away from the windows and hallways in their home. - PKI and digital certificates. PKI can secure client-server connections between multiple networked devices. Using a two-key asymmetric cryptosystem, PKI can facilitate the encryption and decryption of private messages and interactions using digital certificates. These systems help to protect the clear text information input by users into websites to complete private transactions. E-commerce wouldn't be able to operate without the security of PKI.
- Network security. Networks provide a huge opportunity for threat actors to remotely control IoT devices. Because networks involve both digital and physical components, on-premises IoT security should address both types of access points. Protecting an IoT network includes ensuring port security, disabling port forwarding and never opening ports when not needed; using antimalware, firewalls, intrusion detection systems and intrusion prevention systems; blocking unauthorized IP addresses; and ensuring systems are patched and up to date.
![Various components of IoT security architecture]()
Protecting the network is a key responsibility of IoT security. - API security. APIs are the backbone of most sophisticated websites. They enable travel agencies, for example, to aggregate flight information from multiple airlines into one location. Unfortunately, hackers can compromise these channels of communication, making API security necessary for protecting the integrity of data being sent from IoT devices to back-end systems and ensuring only authorized devices, developers and apps communicate with APIs. T-Mobile's 2018 data breach exposed the consequences of poor API security. Due to a leaky API, the mobile giant exposed the personal data of more than 2 million customers, including billing ZIP codes, phone numbers and account numbers.
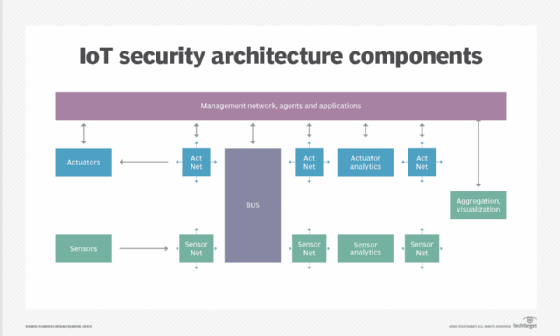
Additional IoT security methods
Other ways to introduce IoT security include the following:
- Network access control (NAC). NAC can help identify and inventory IoT devices connecting to a network. This provides a baseline for tracking and monitoring devices.
- Segmentation. IoT devices that need to connect directly to the internet should be segmented into their own networks and have restricted access to the enterprise network. Network segments should monitor for anomalous activity, taking action if an issue is detected.
- Security gateways. Acting as an intermediary between IoT devices and the network, security gateways have more processing power, memory and capabilities than the IoT devices themselves, which lets them add features such as firewalls to ensure hackers can't gain access to the IoT devices they connect.
- Patch management and continuous software updates. It's critical to provide a way to update devices and software either over network connections or through automation. Having a coordinated disclosure of vulnerabilities is also important for updating devices as soon as possible. Consider end-of-life strategies as well.
- Training. IoT and operational system security are new to many existing security teams. Security staff must keep up to date with new or unknown systems, learn new architectures and programming languages, and be ready for new security challenges. C-level and cybersecurity teams should receive regular cybersecurity training to keep up with modern threats and security measures.
- Team integration. Along with training, integrating disparate and regularly siloed teams can be useful. For example, having programming developers work with security specialists can help ensure the proper controls are added to devices during the development phase.
- Consumer education. Consumers must be made aware of the dangers of IoT systems and provided steps to stay secure, such as updating default credentials and applying software updates. Consumers can also play a role in requiring device manufacturers to create secure devices and refusing to use those that don't meet high-security standards.
- Enforcement and automation of zero-trust policies. The zero-trust model dictates that all users -- whether inside or outside the organization's network -- must be verified, authorized and continually evaluated for security configuration and posture before being given access to applications and data. Automating zero-trust policies and enforcing them across the board can help mitigate security threats against IoT devices.
- Multifactor authentication (MFA). MFA adds an extra layer of security by requiring more than one form of identification when requesting access to a device or network. By enforcing MFA policies, both enterprises and home users can improve the security of IoT devices.
- Machine learning (ML). ML technology can be used to secure IoT devices by automating the management and scanning of devices throughout the entire network. Since every device connected to the network is scanned, it stops assaults automatically before IT teams are alerted. That's what happened in 2018 when Microsoft Windows Defender software stopped a Trojan malware attack in 30 minutes.
Which industries are most vulnerable to IoT security threats?
IoT security hacks can happen anywhere -- from a smart home to a manufacturing plant to a connected car. The severity of the attack depends greatly on the individual system, the data collected and the information it contains.
For example, an attack disabling the brakes of a connected car or hacking a connected health device, such as an insulin pump, can be life-threatening. Likewise, an attack on a refrigeration system housing medicine that's monitored by an IoT system can ruin the viability of a medication if temperatures fluctuate. Similarly, an attack on critical infrastructure, such as an oil well, energy grid or water supply, can be disastrous.
Other attacks, however, can't be underestimated. For example, an attack against smart door locks could potentially allow a burglar to enter a home. Or, in other security breaches, an attacker could pass malware through a connected system to scrape personally identifiable information, wreaking havoc for those affected.
Generally speaking, industries and agencies that are most vulnerable to IoT security threats include, but aren't limited to, the following:
- Retail companies.
- Trucking industry.
- Consumer electronics.
- Utilities and critical infrastructure.
- Healthcare.
- Education.
- Government agencies.
- Financial institutions.
- Energy and utility companies.
Which IoT devices are most vulnerable to security breaches?
In a home-based setting, typically, IoT devices such as smart TVs, refrigerators, coffee machines and baby monitors are known to be vulnerable to security attacks.
In enterprise settings, medical equipment and network infrastructure devices, such as video cameras and printers, can be potential targets. According to research from IoT security provider Armis, 59% of the IP cameras their platform monitored in clinical settings have critical severities, while the second-most dangerous IoT equipment in clinical sites are printers, which have 37% unpatched Common Vulnerabilities and Exposures, 30% of which are critical severity.
Notable IoT security breaches and IoT hacks
Security experts have warned of the potential risk of large numbers of insecure devices connected to the internet since the IoT concept first originated in the late 1990s. Many attacks subsequently have made headlines -- from refrigerators and TVs being used to send spam to hackers infiltrating baby monitors and talking to children. Many IoT hacks don't target the devices themselves, but rather use IoT devices as an entry point into the larger network.
Notable IoT security attacks include the following:
- In 2010, researchers revealed that the Stuxnet virus was used to physically damage Iranian centrifuges, with attacks starting in 2006 but the primary attack occurring in 2009. Often considered one of the earliest examples of an IoT attack, Stuxnet targeted supervisory control and data acquisition systems in industrial control systems, using malware to infect instructions sent by programmable logic controllers. Attacks on industrial networks have continued, with malware such as CrashOverride -- also known as Industroyer -- Triton and VPNFilter targeting vulnerable operational technology and IIoT systems.
- In December 2013, a researcher at enterprise security firm Proofpoint Inc. discovered the first IoT botnet. According to the researcher, more than 25% of the botnet was made up of devices other than computers, including smart TVs, baby monitors and household appliances.
- In 2015, security researchers Charlie Miller and Chris Valasek executed a wireless hack on a Jeep, changing the radio station on the car's media center, turning its windshield wipers and air conditioner on, and stopping the accelerator from working. They said they could also kill the engine, engage the brakes and disable the brakes altogether. Miller and Valasek were able to infiltrate the car's network through Chrysler's in-vehicle connectivity system, Uconnect.
- Mirai, one of the largest IoT botnets to date, first attacked journalist Brian Krebs' website and French web host OVH in September 2016; the attacks clocked in at 630 gigabits per second and 1.1 terabits per second, respectively. The following month, domain name system service provider Dyn's network was targeted, making a number of websites, including Amazon, Netflix, Twitter and The New York Times, unavailable for hours. The attacks infiltrated the network through consumer IoT devices, including IP cameras and routers. A number of Mirai variants have since emerged, including Hajime, Hide 'N Seek, Masuta, PureMasuta, Wicked and Okiru.
- In a January 2017 notice, the Food and Drug Administration warned that the embedded systems in radio frequency-enabled St. Jude Medical implantable cardiac devices -- including pacemakers, defibrillators and resynchronization devices -- could be vulnerable to security intrusions and attacks.
- In July 2020, Trend Micro discovered an IoT Mirai botnet downloader that was adaptable to new malware variants, which would help deliver malicious payloads to exposed Big-IP boxes. The samples found also exploited recently disclosed or unpatched vulnerabilities in common IoT devices and software.
- In March 2021, security camera startup Verkada had 150,000 of its live camera feeds hacked by a group of Swiss hackers. These cameras monitored activity inside schools, prisons, hospitals and private company facilities, such as Tesla.
- In late 2022, hackers began exploiting a series of 13 IoT vulnerabilities related to remote code execution. They installed a modified version of the Mirai malware on compromised devices, giving them unauthorized control over the affected systems.
- In March 2023, Akuvox's smart intercom was found to have zero-day flaws that allowed remote eavesdropping and surveillance.
- Also in March 2023, vulnerabilities in the Trusted Platform Module 2.0 protocol related to buffer overflow were found, putting billions of IoT devices at risk.
IoT security standards and legislation
Many IoT security frameworks exist, but there's no single industry-accepted standard to date. However, simply adopting an IoT security framework can help; they provide tools and checklists to help companies that are creating and deploying IoT devices. Such frameworks have been released by the nonprofit GSM Association, IoT Security Foundation, Industry IoT Consortium and other organizations.
Other IoT security standards and regulations include the following:
- In September 2015, the Federal Bureau of Investigation released a public service announcement, FBI Alert Number I-091015-PSA, which warned about the potential vulnerabilities of IoT devices and offered consumer protection and defense recommendations.
- In August 2017, Congress introduced the IoT Cybersecurity Improvement Act, which would require any IoT device sold to the U.S. government to not use default passwords, not have known vulnerabilities and offer a mechanism to patch the devices. While aimed at those manufacturers creating devices being sold to the government, it set a baseline for security measures all manufacturers should adopt.
- While not IoT-specific, the General Data Protection Regulation, released in May 2018, unifies data privacy laws across the European Union. These protections extend to IoT devices and their networks.
- In June 2018, Congress introduced the State of Modern Application, Research and Trends of IoT Act (SMART IoT Act) to propose the Department of Commerce to conduct a study of the IoT industry and provide recommendations for the secure growth of IoT devices. While the SMART IoT ACT hasn't been passed into law yet, it's been introduced in multiple sessions of Congress.
- In September 2018, the California state legislature approved Senate Bill 327 Information privacy: connected devices, a law that introduced security requirements for IoT devices sold in the U.S.
- In February 2019, the European Telecommunications Standards Institute released the first globally applicable standard for consumer IoT security -- an area that previously hadn't been addressed on such a scale.
- In January 2020, the Developing Innovation and Growing the Internet of Things Act, or DIGIT Act, passed the Senate. This bill requires the Department of Commerce to convene a working group and create a report on IoT, including security and privacy.
- In December 2020, former President Donald Trump signed the IoT Cybersecurity Improvement Act of 2020, directing the National Institute of Standards and Technology to create minimum cybersecurity standards for those IoT devices controlled or owned by the U.S. government.
- In 2022, the U.K.'s Product Security and Telecommunications Infrastructure Act went into effect. This law requires all consumer smart devices to be able to mitigate and protect against cyber attacks.
IoT attacks and security varies
IoT security methods vary depending on the specific IoT application and its place in the IoT ecosystem. For example, IoT manufacturers -- from product makers to semiconductor companies -- should concentrate on building security into their devices from the start, making hardware tamperproof, building secure hardware, ensuring secure upgrades, providing firmware updates and patches, and performing dynamic testing.
Developers of IoT devices should focus on secure software development and secure integration. For those deploying IoT systems, hardware security and authentication are critical measures. Likewise, for operators, keeping systems up to date, mitigating malware, auditing, protecting infrastructure and safeguarding credentials are key. With any IoT deployment, it's critical to weigh the cost of security against the risks prior to installation, however.
IoT endpoints have emerged as top targets for cybercriminals. Discover the top 12 IoT security threats and how to prioritize them.







